The Drug Development Based on Pathogenetic Research in Alzheimer’s Disease ()
1. Introduction
Alzheimer’s disease (AD) is described as one of the most common neurodegenerative disorders, with a prevalence of 5 percent after sixty-five years of age, increasing to almost 30 percent in people over age eighty-five [1] . The clinical symptoms of AD include loss of memory, progressive cognitive impairment, various behavioral disturbances and neurological disorders. Typically, AD start with mild memory deficits, then gradually progress to severe dementia and stupor. Generally speaking, about nine years after clinical diagnosis, the AD patients may face to death caused by respiratory complications [2] . Neuropathologically, AD is characterized by senile plaques (SP) composed by amyloid-β peptides (Aβ), neurofibrillary tangles (NFTs) generated by hyper-phosphorylated forms of protein tau [3] and degeneration or atrophy of the basal forebrain cholinergic neurons.
APP is overexpressed in AD [4] . After the cleavage of APP by two proteases, denoted as β-secretase (BACE1) and γ-secretase, the sequential action will aggregates into toxic Aβ. The two major subtypes are Aβ1-40 and Aβ1-42. The shorter one comes from the cutting of typical APP in endoplasmic reticulum, while the longer one is formed across the Golgi network. Compared with each other, Aβ1-40 is more common, but Aβ1-42 is even more relevant to the disease. Tau is a component of microtubules; it stabilizes growing axons and is necessary for neuritis [4] . In AD, tau is abnormally hyperphosphorylated and forms insoluble fibrils, which contribute to the earliest cytoskeletal changes in NFTs formation. This abnormal spiral structure interfere the normal function of neurons, ultimately leading to neuronal cell death. Cholinesterase is the key enzyme in biological nerve conduction. In the cholinergic synaptic cleft, it degrades acetylcholine, induces the termination of excitability role of the neurotransmitter on postsynaptic membrane, which ensures normal convery of nerve signals in vivo. Acetylcholinesterase catalyzes the cleavage reaction of acetylcholine, resulting in the lack of acetylcholine, thus interfere nerve signal transmission.
Alzheimer’s disease pathology changes major involve cholinergic nerve pathways from frontal base to the cerebral cortex and hippocampus. As we all known, these pathways are associated with attention, learning ability, memory and other cognitive processes. The loss of basal forebrain cholinergic cells in AD patients leads to reduction of synaptic availability of acetylcholine. So, AD patients always suffer the cognitive impairment.
Scientists have been working on the pathophysiological processes of Alzheimer’s disease for more than a century. A large number of theories have been discovered to explain what is happening in the brain of AD patients. For example, Aβ causes calcium dysregulation and oxidative stress in central nervous system cells, inflammatory changes can be observed in the brain, even diabetes and insulin-resistance may have connection with AD through GSK3β. Nevertheless, there is a long way to go before we discover the exactly and comprehensive functions of Aβ and tau in the process of AD. In other hand, the significantly effective drugs for AD clinical treatment are less than enough. In this article, we focus on the molecular and cellular alterations involved in neuronal dysfunctions caused by Aβ and tau in AD. More importantly, the drug development based on pathogenetic research in AD, and possible research priorities in the future will be mentioned.
2. Cholinergic Drugs
Neurochemical research shows that the brain in AD patients have obvious shortage of central cholinergic neurotransmitter, causing loss of memory, directional force, behavior and personality change as a result. Enhancing cholinergic effects is an important way for the treatment of AD. Current research focuses on acetylcholinesterase inhibitors (AChEI), which can increase ACh concentration in the synaptic cleft by reducing its degradation, thus improve the central activity of choline. It is by far the most commonly used and is considered to be one of the most promising drug treatments of AD. The second generation of AChEI is widely applied in clinical treatment, that is Donepezil hydrochloride (aricept) [5] , Rivastigmine Tartrate (exelon) [6] , Galantamine and Huperzine A.
Donepezil hydrochloride is a highly selective AChEI. It is approved for use in mild-to-moderate AD patients [7] [8] . It has shown some benefit in slowing hippocampal atrophy and protecting nerve cells. Rivastigmine Tartrate has easy BBB permeability and is approved for mild-to-moderate AD [9] . Rivastigmine Tartrate can selectively enhance the effect of acetylcholine in cerebral cortex and hippocampus. Furthermore, cholinesterase inhibitors can slow down the the formation of the amyloid precursor protein (APP) fragment. After combining with its target enzyme into covalent compounds, Rivastigmine Tartrate causes a temporary loss of activity of the enzyme. Galantamine has allosteric nicotinic receptor modulation properties. Galantamine may increase ACh release by regulating brain external nicotinic acid receptor. Research showed that galantamine may be safe for the treatment of elderly patients with severe AD, which improves cognitive function in patients. However, everyday life parameters change is not obvious. Another ChEI, Huperzine A, is considered to be one of the treatment for memory disorders [10] . However, for its lacking of proprietary patent for the treatment of AD, it is considered as a nutraceutical supplement in the US. Clinical and preclinical toxicities are to be established in the future.
AD is a multifactorial disease, so the innovative model is to achieve the goal of “one molecule, multiple targets”. Ideally, hybrids can provide parent compounds more potency such as BBB permeability, additional receptors or epitopes [11] . Human studies are planned.
3. Calcium Regulation as a Therapeutic Approach for AD
There is considerable evidence that Aβ induces calcium dysregulation in neuron. Sequential cleavages of APP by secretases generate sAPPα, Aβ and AICD. Intracellular APP domain (AICD) can modify nuclear gene transcription and further more perturb Ca2+ homeostasis. On the other hand, sAPPα, generated from APP by α-secretase, is normally produced to active K+ channels, thereby hyperpolarizing the membrane and reducing Ca2+ influx [12] . The production of sAPPα plays a protecting role in neurons; however, amyloidgenic processing may prevent this program. Aβ oligomers enhance calcium ion influx by Ca2+-permeable channels, which can be facilitated by binding to phosphatidylserine (PtdS). Cell-surface exposure of PtdS is usually indicative of apoptotic cells. Mitochondrial, the energy supply station of cells, once effected by Aβ, will induce superoxide anion radical (O2•−) production, Ca2+ overload, and decreased ATP production. In this condition, PtdS will flip from the inner portion to the cell surface of the plasma membrane. In turn, neurons with lower ATP level are particularly susceptible to Aβ toxicity [13] . Aβ can also generate hydroxyl radical (OH) in the presence of Fe2+ and Cu+ [14] [15] . As a result, the function of ATPases dependent ion channels (Na+ and Ca2+ pumps) can be impaired by toxic aldehydes generated by membrane lipid peroxidation (LP). Therefore, the membrane becomes depolarized and toxic amounts of Ca2+ flux into the cytoplasm through the open channels, glutamate receptor channels (N-methylD-aspartate receptor, NMDAR) and voltage-dependent Ca2+ channels (VDCC) open. Inside the neurons, Presenilins (PS) functions as an endoplasmic reticulum (ER) Ca2+ leak channel. In familial Alzheimer’s disease, PS mutations cause excessive accumulation of Ca2+ and then enhance Ca2+ release by inositol 1, 4, 5-trisphosphate receptors (IP3R) channels and ryanodine receptor (RyR). Aβ can also block the response of nicotinic acetylcholine receptors (nAChRs) and induces sustained Ca2+ levels increases in presynaptic through IP3 [16] (Figure 1).

Figure 1. Molecular and cellular alterations involved in neuronal dysfunction in AD. The amyloid-β precursor protein (APP) can be cleaved by β-secretase and γ-secretase, resulting in the liberation of sAPPα, the amyloid-β peptitde (Aβ) and intracellular APP domain (AICD). AICD can translocate to the nucleus and perturb Ca2+ homeostasis. sAPPα may active K+ channels. Aβ oligomers enhance calcium ion influx into the cell by the formation of Ca2+-permeable channels. Aβ will also induce superoxide anion radical (O2•−) production, Ca2+ overload, and decreased ATP production in mitochondrial. Aβ can also interact with Fe2+ and Cu+ to generate hydrogen peroxide and hydroxyl radical (OH). As a result, the function of Na+ and Ca2+ pumps can be impaired by toxic aldehydes generated by membrane lipid peroxidation (LP), resulting the Ca2+ flux through N-methyl-D-aspartate receptor (NMDAR) and voltage-dependent Ca2+ channels (VDCC). In familial Alzheimer’s disease, Presenilins (PS) mutations cause excessive accumulation of Ca2+ in the endoplasmic reticulum (ER) and then enhance Ca2+ release through inositol 1,4,5-trisphosphate receptors (IP3R) channels and ryanodine receptor (RyR). Aβ can also induce sustained nAChR-mediated increases in presynaptic Ca2+ levels through IP3.
Inspired by the above theory, methods to stabilize neural intracellular calcium homeostasis may be one of the treatments for AD. So far, a variety of drugs have therapeutic potential in vivo or in vitro experiments.
Since excessive activation of glutamate system causes excitotoxic neuronal death, NMDA open channel blocker, can antagonize glutamate excitotoxicity. Memantine, which belongs to NMDA receptor antagonist, is different from the cholinesterase inhibitor. It is the first and only drug that is approved for treatment of moderate to severe AD. Clinical studies have demonstrated that it benefits in cognitive and behavioral outcomes in patients, either as monotherapy or in combination with donepezil [17] . It also shows good effect to main types of dementia: Alzheimer’s dementia, vascular dementia and AIDS dementia, which indicate that it has an advangtage over other kinds of AD drugs on the market. Animal experiments showed that Nimodipine, an L-type VGCC inhibitor, plays a role as calcium antagonist. It decreases the intracellular calcium ion concentration, promotes the regeneration of injured neurons, enhance the plasticity of the aging central nervous system. In the clinical treatment, Nimodipine has obvious curative effect on memory impairment due to AD [18] . Recently, nimodipine was reported to selectively stimulate secretion of Aβ1-42 slightly [19] . But the exact mechanism has not yet been elucidated.
In vitro experiments, ultra-low-molecular-weight heparin (ULMWH) partly reduced the [Ca2+]i increase induced by glutamate, this suggests that ULMWH may inhibit external Ca2+ influx mediated by NMDA receptor. In addition, the IP3R induced [Ca2+]i was significantly suppressed ULMWH, suggests that ULMWH can regulate calcium balance by inhibiting calcium ion release. So ULMWH may be speculated as a specific IP3R antagonist just like heparin. In this way, less IP3 binds to IP3Rs and the increase of [Ca2+]i was blocked. Therefore, factors such as ULMWH are expected to have good effect on AD.
4. Anti-Amyloid Therapies
As Aβ is the heart of the amyloid hypothesis of AD, the formation of Aβ oligomers is a directly AD killer. The mainly function of β-secretase and γ-secretase is to produce toxic Αβ1-42. Currently the focus of scientists is the inhibition of activity of β-secretase and γ-secretase. β-secretase (BACE1) initiates the amyloidogenic pathway. Activation of nuclear peroxisome proliferator-activated receptor γ (PPARγ) can suppress expression of β-secretase. Rosiglitazone and pioglitazone, which belong to oral drugs for type 2 diabetes, can act as β-secretase inhibitors by stimulating PPARγ [20] . The therapeutic effects of PPARγ agonists in AD may be caused by their effect of increasing insulin sensitivity and reducing concentrations of insulin.
Present study is generally believed that insulin can not only pass through the blood brain barrier, but also be synthesized within the brain tissue. The insulin receptor (IR) and receptor signal transduction molecules exist in brain tissue [21] . Insulin can not only regulate sugar metabolism and energy metabolism, but also have various biological functions. The function of brain insulin is complex, including supporting the surviving of mature neurons. Cascade control apoptosis is one of the important roles of insulin. The common pathological mechanism of diabetes encephalopathy and AD is an obstacle of insulin signal transduction pathways, namely the insulin resistance, which can cause metabolic disorders and cognitive dysfunction. In a word, insulin signaling induces the phosphorylation and inhibition of glycogen synthase kinase 3 (GSK3). It would therefore promote tau phosphorylation, leading to aggregation and tangle formation, as well as contributing to Aβ peptide production and plaque formation [22] (Figure 2). The related content remains further research.
Using Αβ antibody in treatment of AD is a current research hot spot. Active and passive immunization clinical research is ongoing in several pharmaceutical companies. According to animal tests, vaccination of synthetic Αβ1-42 immunization can produce Αβ1-42 antibody, thus causes activation of monocyte or microglia. As a result, immune therapy has become a new method for treatment of AD, which has entered clinical trials in the United States. Preclinical and early clinical trial results show Αβ immunotherapy has great potential to overcome the AD. Active immunizations include injection of synthetic Αβ peptide, Αβ fragment joined with carrier protein or adjuvant. It will stimulate the host to produce antibodies against Αβ. Passive immunization is to directly inject Αβ specific antibody into the host, thus activate the host immune system. The common basic principle of active and passive immunotherapy is removing Αβ from the brain. In clinical trials of AN-1792 vaccine, the number of Αβ has decreased in the brains of patients. However, some patients appeared subacute aseptic meningoencephalitis within Ⅱ period clinical trials [23] . Better than AN-1792 vaccine, CAD-106 vaccine, did not show such side effect in the early human trials [24] . A new strategy to increase the security of active immunization is to optimize the drug delivery way. It has been proved that, intranasal administration of Αβ peptide, without adjuvant,
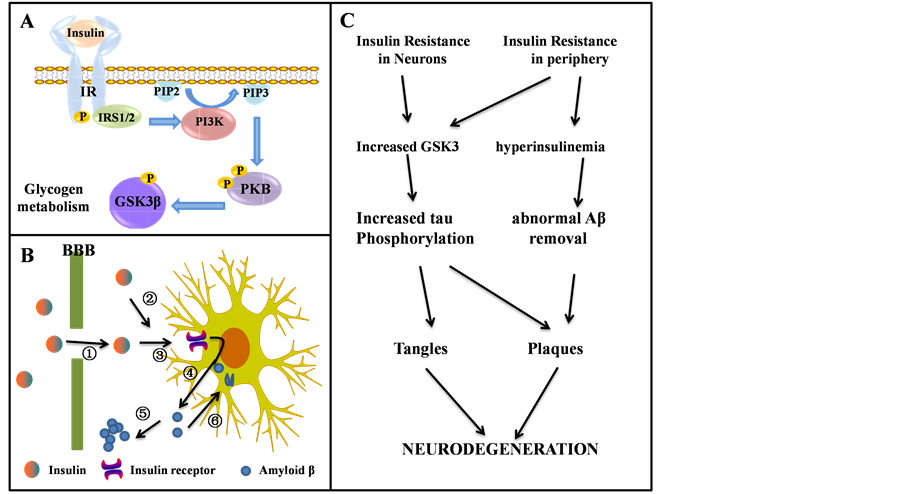
Figure 2. The role of insulin receptor (IR) in neurodegeneration, which is expressed in both neurons and glia. (A) Schematic representation of insulin signaling; (B) The potential role of insulin in the pathogenesis of AD. Insulin is transported actively across the blood-brain barrier (①), it can also be produced locally in the brain (②). Insulin act through cerebral insulin receptors (③). Thus stimulates the secretion of Aβ into the extracellular space (④) where it can aggregate into senile plaques (⑤). Alternatively, excessive Aβ can be cleared through endocytosis (⑥); (C) The connection between insulin resistance and neurodegeneration.
leads to appropriate immunoreaction. Gene immunotherapy, a kind of active immunization in essence, has much potential. Initially, Aβ-DNA vaccine was coded by adenovirus or adeno-associated virus vector. Researchers are focusing on the development of non-viral plasmid vector, because of the large-scale low-cost production and no risk of infection or transfection [25] .
As for passive immunization, many monoclonal antibody is in development and testing, such as Bapineuzumab (AAB-001) [26] and Solanezumab (LY2062430) [27] [28] . But the production of monoclonal antibodies cost great and need repeated injection. What’s worse, it is hard to choose the appropriate target and pass through the BBB. It also has a risk of bleeding.
5. Drugs to Target Tau Protein
Tau protein is a kind of microtubule associated protein whose primary function is to stabilize the conformation of microtubules (MTs). The phosphorylation level of tau protein in AD patients’ brain is three times higher than normal. Excessive phosphorylated tau protein may dissociate from microtubules and aggregates into nerve toxic NFTs [29] . As a consequence, microtubule is disintegrated and cytoskeleton is damaged, too.
There are two main therapeutic approaches aiming at tau protein: inhibitors of phosphorylase kinase of tau protein and compounds that inhibit tau aggregation or promote its disassembly. Inhibition of the excessive phosphorylation of tau protein is the major research direction for the treatment of AD. GSK3, and specifically its beta isoform (GSK3β), have been shown to closely connected with phosphorylate tau [30] . Animal studies have confirmed that in the early pathological changes of tao protein, lithium salt, one kind of non-specific GSK3 inhibitor [31] , can prevent excessive phosphorylation of tau protein and block the further progress of the disease. But lithium salt has little effect during the late stage, so researchers have turned to develop specific inhibitors of phosphorylase kinase of tau protein. According to the research results, some kind of small molecular compounds can prevent the interaction of tau protein, thus inhibit its aggregation. For example, methylene blue, a widely used histology dye, can not only prevent polymerization of tau protein into oligomers, but also reduce Aβ level in the brain [32] . Methylene blue also has antioxidant properties and was effective in improving learning deficits either used alone or in combination with rivastigmine in animals [33] . So it is considered to be a promising new AD treatment. It has a high bioavailability when tested in a phase 2 study in patients with moderate AD [34] and its safety and clinical efficacy need to be further confirmed.
In addition to the two kinds of medicine mentioned above, new drugs designed on counteracting the functional loss of tau protein are promising. As we all know, the primary function of tau protein is to stabilize the conformation of MTs. Over the past decades, several classes of MT-stabilizing products have been used as antineoplastic drug. However, people are worried about dose-limiting toxicities if this class of therapeutics is used in long-term treatment of tauopathy patients. Paclitaxel, which belongs to taxanes natural products, binds to the lumen of the MT at β-tubulin subunit [35] . Importantly, paclitaxel is found to having a function of promoting MT stabilization instead of tau protein. [36] . The actin mode of epothilones is similar to paclitaxel. In vitro, both paclitaxel [37] -[41] and epothilones [42] -[44] have been found to protect neurons against neurotoxicity mediated by tau protein or Aβ. However, paclitaxel could not cross the BBB while epothilone D shows to be a brain-penetrant MT-stabilizing agent [45] . Considering that tau pathology is primarily in the brain, only epothilone D can be suitable as a therapeutic candidate for human tauopathies. As to the consideration of dose-limiting toxicities, one important observation made in vivo studies in epothilone D [46] indicated that low doses of epothilone D may produce optimal therapeutic effects. Overstabilization of MTs by high dose of agents on the other hand become counterproductive and may be accompanied by side effects. As a growing number of MT-stabilizing products are being discovered, a particular attention should be paid to these agents to find more useful treatment for AD and other tauopathies.
6. Summary and Prospective
The study of the pathophysiological processes of AD has been attracting people’s attention for more than a century. Aβ and NFTs have been observed to have extensively damage to normal function of central nervous system, such as cholinergic deficit in the CNS, intracellular Ca2+ disequilibrium, inordinate oxidative stress and inflammatory processes. These pathophysiological processes of AD provide multifarious targets for therapeutic or preventative agents. Besides those five drugs which are currently ratified for use in the treatment of AD (donepezil (Aricept®), galantamine (Reminyl®), rivastigmine (Exelon®), tacrine (Cognex®) and memantine (Namenda®)), many other kinds of drugs aiming at processes mentioned above have potentials in AD treatment, for example, GAG mimetic, NSAIDs, and hypoglycemic agents (Table 1).
According to the past failures we have met in AD drug clinical trials that were conducted over the last decades, we can come to the conclusion that it is questionable to conducting large clinical development programs in AD blindly. AD is the result of the interaction of pathogenic factors. It is necessary to be better appreciating the complicacy of the disease. In our way to find significant treatment for AD, it is important to better understand the relationship between tau, Aβ and other factors. In a word, the competing risk factors, physiological factors such as age and genetics, and environmental factors all play non-ignorable roles in the progress of AD. As a result, drugs with wide range of targets always appear little effect in the treatment.
The failures in AD drug clinical trials also forced people to turn to another approach to the treatment of AD. There are three kinds of behaviors that are considered to reduce the risk of AD [56] . Those are cognitive stimu

Table 1. Different classes of products and their stages of development as potential candidates for AD.
RCT: Randomized Controlled Trial. MCI: Mild Cognitive Impairment.
lation, mental and physical exercise, and dietary energy restriction. They are thought to show beneficial effect by activating neurotransmitters, growth factors, and hormone receptors. In this way, the synaptic activity in nerve cell networks is increased and neurons will be protected against oxidative and metabolic stress.
What’s more, most compounds showed some benefits in mild AD, either than moderate AD. These discoveries inspire us that the early we carry out therapeutic trials, the more possible it will be to block the course of the disease. As a consequence, the identification of more accurate tools for early diagnosis is needed. If we have new markers that can detected in the blood or other body fluids at the mild phase of AD, it will be a useful aid for the diagnosis and management of patients with AD.
Acknowledgements
The authors thank Dr. Li-Na Hao for her insightful advice, and Ms. Yu-Ning Song for her assistance in preparing the manuscript.